2022. aasta uus ravimiülevaade
USA Pharm. 2022;47(10):24–30.
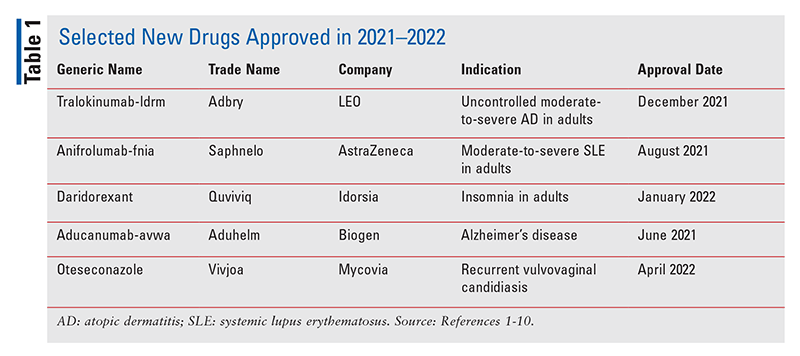
Uued molekulaarsed üksused (NME-d), nagu FDA on määratlenud, on uued ravimtooted, mis sisaldavad toimeainena keemilist ainet, mida turustatakse esmakordselt Ameerika Ühendriikides. Järgmised aastatel 2021–2022 heaks kiidetud NME-de kirjeldused ( TABEL 1 ) kirjeldab üksikasjalikult iga uue ravimi kliinilisi ja farmakoloogilisi põhiprofiile ning peamisi ettevaatusabinõusid ja hoiatusi. Samuti on lisatud lühike kokkuvõte valitud farmakokineetilistest, kõrvaltoimetest, ravimite koostoimetest ja annustamisandmetest, mis on FDA-le esitatud tootja uue ravimitaotluse toetuseks. See ülevaade on pigem objektiivne kui sisult hindav. Teave iga NME kohta saadi peamiselt allikatest, mis avaldati enne FDA heakskiitu. Kogemused näitavad selgelt, et NME terapeutilise profiili paljusid aspekte ei tuvastata turustamiseelsetes kliinilistes uuringutes ja need ilmnevad pärast ravimi kasutamist laiemas elanikkonnas. Näiteks ilmnevad varem teatamata kõrvaltoimed mõne uue turuosalise puhul mitme aasta jooksul pärast nende turustamist. Mõned NME-d võivad lõpuks hankida vähemalt ühe musta kasti hoiatuse ravimi tõsiste kõrvaltoimete kohta või nad kõrvaldatakse turult ohutuse põhjustel, mida heakskiitmise ajal ei tunnustatud. Seega, kuigi käesolev ülevaade tutvustab mõningaid uusi ravimeid, on oluline, et praktikud oleksid teadlikud muutustest nende raviprofiilides, nagu on kirjeldatud farmaatsiaalases kirjanduses ja patsientide poolt.
Tralokinumab-ldrm (Adbry, LEO)
Näidustused ja kliiniline profiil 1.2 : FDA kiitis tralokinumab-ldrm-i heaks mõõduka kuni raske atoopilise dermatiidi (AD) raviks täiskasvanutel, kelle haigus ei allu paiksete ravimitega hästi või kui need ravimeetodid ei ole soovitatavad. AD, tuntud ka kui atoopiline ekseem , on krooniline korduv põletikuline nahahaigus, mida iseloomustab naha kuivus, sügelus (eriti öösel) ja punased kuni pruunikashallid laigud erinevatel kehaosadel. AD esinemissagedus on tööstusriikides kasvanud peaaegu kolm korda, mõjutades maailmas umbes 15–20% lastest ja 1–3% täiskasvanutest. Tralokinumab-ldrm, esimene AD raviks heaks kiidetud bioloogiline aine, inhibeerib interleukiini (IL)-13, mis on selle haiguse peamine põletikuline vahendaja. Tralokinumab-ldrmi võib kasutada koos paiksete kortikosteroididega või ilma.
Selle ravimi heakskiitmine põhines kolme topeltpimeda, platseebokontrolliga, rahvusvahelise III faasi uuringu (ECZTRA 1, ECZTRA 2 ja ECZTRA 3) efektiivsuse ja ohutuse tulemustel, mis hõlmasid enam kui 1500 täiskasvanud patsienti. Patsiendid said tralokinumab-ldrm 300 mg süste igal teisel nädalal kas eraldi või koos paiksete kortikosteroididega. Peamised tulemusnäitajad olid Investigator's Global Assessment skoor 0 või 1 (IGA 0/1) 16. nädalal ja ³75% paranemine ekseemi pindala ja raskusastme indeksis (EASI 75) 16. nädalal. Patsiendid, kes saavutasid IGA 0/1 ja/või EASI 75 tralokinumab-ldrm-ga 16. nädalal randomiseeriti tralokinumab-ldrm-iga iga 2 või 4 nädala järel või platseebo rühmaks kuni 36 nädala jooksul. 16. nädalal saavutas IGA 0/1 (38,9% vs 26,2%) ja EASI 75 (56,0% vs 35,7%) rohkem tralokinumab-ldrm patsiente kui platseeboga patsiente. 16. nädalal ravile reageerinutest säilitas IGA 0/1 ja EASI vastavalt 89,6% ja 92,5% neist, keda raviti tralokinumab-ldrm-iga iga 2 nädala järel ning 77,6% ja 90,8% neist, keda raviti tralokinumab-ldrm-iga iga 4 nädala järel. 75 32. nädalal. Patsientide seas, kes ei saavutanud tralokinumab-ldrm-iga 16. nädalal IGA 0/1 ja EASI 75, saavutasid need tulemusnäitajad 32. nädalal vastavalt 30,5% ja 55,8%.
Farmakoloogia ja farmakokineetika 1.2 : Tralokinumab-ldrm on inimese immunoglobuliini (Ig) G4 monoklonaalne antikeha, mis seondub spetsiifiliselt inimese IL-13-ga ja inhibeerib selle koostoimet selle retseptori alfa-1 ja alfa-2 subühikutega. IL-13, tsütokiin, mida tavaliselt toodab 2. tüüpi immuunvastus, soodustab AD-ga seotud põletikueelsete tsütokiinide, kemokiinide ja IgE vabanemist.
Tralokinumab-ldrm-i absoluutne biosaadavus on hinnanguliselt 76% ja aeg saavutab maksimaalse plasmakontsentratsiooni (T max ) 5–8 päeva pärast manustamist. Jaotusruumala on hinnanguliselt 4,2 l. Tralokinumab-ldrm näib läbivat peptiidide metabolismi. Selle poolväärtusaeg on 3 nädalat ja süsteemne kliirens on hinnanguliselt 0,149 l/päevas.
Kõrvaltoimed ja ravimite koostoimed 1.2 : Tralokinumab-ldrm-iga kliinilistes uuringutes teatatud sagedasteks kõrvaltoimeteks olid eosinofiilia, silma- ja silmalaugude põletik, ülemiste hingamisteede infektsioonid ja süstekoha reaktsioonid. Patsientidel soovitatakse teavitada oma tervishoiuteenuse osutajat uutest või süvenevatest silma sümptomitest, nagu konjunktiviit või keratiit. Esinenud on tõsiseid ülitundlikkusreaktsioone, sealhulgas anafülaksia ja angioödeemi, mis on nõudnud ravi katkestamist. Tralokinumab-ldrm võib kahjustada antihelmintiaravi tõhusust; seetõttu ei tohi seda alustada ega jätkata patsientidel, kellel on juba olemasolev helmintiinfektsioon või kes nakatuvad ravi ajal. Tralokinumab-ldrm-ravi ajal tuleb vältida elusvaktsiinide kasutamist.
Mis puudutab ravimite koostoimeid, siis väikeses kliinilises uuringus, milles hinnati tralokinumab-ldrmi mõju midasolaami (CYP3A4 substraat), varfariini (CYP2C9 substraat), omeprasooli (CYP2C19 substraat), metoprolooli (CYP2D6 substraat, YP1A2 (C substraat) ja kofeiini YP1A2 substraat) farmakokineetikale olulisi muutusi kokkupuutes (AUC või C max ) CYP450 substraatide arvu pärast tralokinumabi korduvat manustamist, võrreldes tralokinumab-ldrm monoteraapiaga.
Annustamine ja manustamine 1.2 : Tralokinumab-ldrm on saadaval subkutaanseks süstimiseks 150 mg/ml eeltäidetud süstlas. Soovitatav annus on algannus 600 mg (neli 150 mg süsti), millele järgneb 300 mg (kaks 150 mg süsti), mis manustatakse igal teisel nädalal. Pärast 16-nädalast ravi võib kaaluda 300 mg annust iga 4 nädala järel patsientidele kehakaaluga <100 kg, kui nahk on selge või peaaegu selge. Kõik vanusele vastavad vaktsineerimised, mida soovitavad kehtivad immuniseerimisjuhised, tuleb teha enne ravi alustamist.
Anifrolumab-fnia (Saphnelo, AstraZeneca)
Näidustused ja kliiniline profiil 3.4 : Anifrolumab-fnia on I tüüpi interferooni (IFN) retseptori antagonist, mis on heaks kiidetud mõõduka kuni raske süsteemse erütematoosluupuse (SLE) raviks täiskasvanutel, kes saavad standardravi. Anifrolumab-fnia ohutus ja efektiivsus määrati kindlaks kolmes rahvusvahelises platseebokontrollitud kliinilises uuringus: MUSE, TULIP-1 ja TULIP-2. MUSE oli II faasi uuring, mille esmaste tulemusnäitajate hulka kuulusid SLE ravivastuse indeksi (SRI-4) hindamine ja suukaudsete kortikosteroidide (OCS) pidev vähenemine 24. nädalal. Kokku randomiseeriti 305 patsienti 1:1:1 saama anifrolumaabi. -fnia 300 mg, anifrolumab-fnia 1000 mg või platseebo. III faasi uuringutes TULIP-1 ja TULIP-2 oli haiguse aktiivsuse paranemise esmane tulemus, mida hinnati 52. nädalal, mida mõõdeti SRI-4 TULIP-1 puhul ja Briti saarte luupuse hindamise rühmal põhinev komposiitluupuse hindamine (BICLA) aastal. TULIP-2. TULIP-1 ja TULIP-2 sekundaarsed tulemused hõlmasid OCS-i vähenemise säilitamist, naha SLE aktiivsuse paranemist ja ägenemise määra.
BICLA ravivastuse määrasid 52. nädalal teatati kõigis kolmes uuringus, kuid need olid esmased tulemusnäitajad ainult TULIP-2 puhul. Vastuste määrade erinevus oli MUSE puhul 28,8%, TULIP-1 puhul 17% ja TULIP-2 puhul 16,3%. SRI-4 ravivastuse määr 52. nädalal oli MUSE puhul 24%, TULIP-1 puhul 6% ja TULIP-2 puhul 18,2%. Anifrolumab-fnia ei näidanud statistiliselt olulist erinevust TULIP-1 ravivastuse määrades.
Farmakoloogia ja farmakokineetika 3.4 : Anifrolumab-fnia on immunoglobuliini (Ig) G1-kappa monoklonaalne antikeha, mis seondub I tüüpi IFN-retseptori (IFNAR1) 1. alaühikuga, inhibeerides selle aktiivsust. I tüüpi IFN-ide aktiivsuse vähenemine vähendab nende rolli SLE patogeneesis. Anifrolumab-fnia toimib ka IFNAR1 internaliseerimiseks, vähendades olemasolevaid retseptoreid. Need toimemehhanismid vähendavad põletikku ja immunoloogilisi protsesse, mis soodustavad SLE patoloogiat.
Anifrolumab-fnia farmakokineetika on mittelineaarne annuste vahemikus 100 mg kuni 1000 mg iga 4 nädala järel, mille tulemuseks on suurem AUC ekspositsioon annuse suurendamisel. Jaotusruumala tüüpilisel SLE-patsiendil (69,1 kg) on hinnanguliselt 6,23 l. Standardannuse põhjal oli anifrolumab-fnia kliirens 300 mg IV iga 4 nädala järel hinnanguliselt 0,193 l/päevas.
Kõrvaltoimed ja ravimite koostoimed 3.4 : Kõige sagedasemad (> 10%) anifrolumab-fnia kõrvaltoimed olid infektsioonid (70% patsientidest), sealhulgas gripp, kuseteede infektsioon, tuberkuloos, bronhiit ja (esinemissagedus 34%) ülemiste hingamisteede infektsioonid (st nasofarüngiit, farüngiit). ja sinusiit). Tõsiste infektsioonide esinemissagedus ravi ajal oli 5%. Praegu on andmed anifrolumab-fnia kasutamise kohta inimese raseduse ajal piiratud ja need ei ole piisavad, et anda teavet ravimiga seotud suurte sünnidefektide, raseduse katkemise või ema või loote ebasoodsate tagajärgede riski kohta. On teada, et monoklonaalsed IgG antikehad transporditakse raseduse edenedes aktiivselt läbi platsenta; seetõttu võib loote kokkupuude anifrolumab-fniaga olla suurem kolmandal trimestril.
Ainus teatatud anifrolumab-fnia vastunäidustus on tõsine ülitundlikkus (nt anafülaksia) anifrolumaabi või selle koostise mis tahes komponendi suhtes. Kliiniliselt märkimisväärsed kõrvaltoimed on tõsised infektsioonid, ülitundlikkusreaktsioonid, sealhulgas anafülaksia, ja pahaloomulised kasvajad. Siiani ei ole ametlikke ravimite koostoime uuringuid läbi viidud.
Annustamine ja manustamine 3.4 : Anifrolumab-fnia tarnitakse üheannuselise viaalina, mis sisaldab 300 mg/2 ml (150 mg/ml). Soovitatav annus on 300 mg, mida manustatakse IV infusioonina 30 minuti jooksul. Enne manustamist tuleb anifrolumab-fnia lahjendada 0,9% naatriumkloriidi süstelahusega, USP, kasutades aseptilist tehnikat. 100 ml infusioonikotist eemaldatakse 2 ml 0,9% naatriumkloriidi süstelahust ja õrnalt segades lisatakse 2 ml anifrolumab-fnia, et kogumaht oleks 100 ml. Infusioonilahus tuleb manustada kohe pärast valmistamist 30-minutilise perioodi jooksul läbi infusioonitoru, mis sisaldab steriilset madala valkudega sidumisvõimet 0,2- või 0,22-mikronist filtrit. Pärast infusiooni lõppu tuleb toru loputada 25 ml 0,9% naatriumkloriidi süstelahusega, USP.
Daridoreksant (Quviviq, Idorsia)
Näidustused ja kliiniline profiil 5.6 : Daridoreksant kiideti heaks täiskasvanute unetuse raviks, mida iseloomustavad raskused uinumisel ja/või une säilitamisel. Unetust, kõige levinumat unehäiret (esinevad >30% USA täiskasvanutest), määratletakse kui raskusi piisava unega ja rahulolematust unega koos olulise negatiivse mõjuga päevasele funktsioonile. Kroonilist unetust peetakse raskusteks uinumise alustamisel ja/või une säilitamisel vähemalt 3 ööd nädalas vähemalt 3 kuu jooksul. Unetus, mis näib olevat tingitud ärkvelolekuga seotud ajupiirkondade üleaktiivsusest, mõjutab oluliselt haige tuju, energiataset ja keskendumisvõimet. Lisaks on pikaajaline unetus seotud tõsiste tervisehäiretega, nagu psühhiaatrilised häired, südame-veresoonkonna haigused, II tüüpi diabeet, ainete kuritarvitamine ja dementsus.
Daridoreksandi efektiivsust hinnati kahes võrdselt randomiseeritud, mitmekeskuselises platseebokontrolliga uuringus: uuring 1 ja uuring 2. Kokku osales 1854 unetusega isikut, nagu on määratletud Vaimsete häirete diagnostika ja statistika käsiraamat , 5. väljaanne, sai daridoreksanti või platseebot üks kord päevas õhtuti 3 kuu jooksul. Uuringus 1 määrati 930 isikule 25 mg daridoreksanti, 50 mg daridoreksanti või platseebot; Uuringus 2 määrati 924 isikule 25 mg daridoreksanti, 10 mg daridoreksanti või platseebot. 3-kuulise raviperioodi lõpus hõlmasid mõlemad uuringud 7-päevast platseebo lõppperioodi, mille järel said katsealused osaleda jätkuuuringus. Kokku raviti 600 isikut vähemalt 6 kuud; neist 373 raviti vähemalt 12 kuud. Mõlema uuringu esmased efektiivsuse tulemusnäitajad olid unelaboratooriumis objektiivselt polüsomnograafia abil mõõdetud latentsuse muutus algtasemest 1. ja 3. kuuni (LPS, aeg une esilekutsumiseks) ja ärkamine pärast une algust (WASO, une säilitamine). Uuringus 1 näitasid 25 mg daridoreksanti ja 50 mg daridoreksanti statistiliselt olulist paranemist võrreldes platseeboga LPS ja WASO ning enda teatatud kogu uneaja (sTST) osas 1. ja 3. kuul. 50 mg annus näitas ka päevade märkimisväärset vähenemist. unisus võrreldes platseeboga – mõõdetuna unisuse domeeni skooriga unetuse päevaste sümptomite ja mõjude küsimustikus 7 – 1. ja 3. kuul, mis on peamine teisene tulemusnäitaja. Uuringus 2 näitas 25 mg annus WASO ja sTST statistiliselt olulist paranemist platseeboga võrreldes 1. ja 3. kuul, kuid 10 mg annus mitte.
Farmakoloogia ja farmakokineetika 5.6 : Oreksiin A ja oreksiin B on neuropeptiidid, mida sekreteerivad neuronid, peamiselt lateraalses hüpotalamuses. Need neuropeptiidid kutsuvad esile oma toime, seondudes ja aktiveerides kaks G-valguga seotud retseptorit (GPCR): oreksiini retseptor tüüp 1 ja oreksiini retseptor tüüp 2 (OX1R, OX2R). Oreksiini retseptori rajad mängivad olulist reguleerivat rolli paljudes füsioloogilistes protsessides, sealhulgas une-ärkveloleku rütmis. Daridoreksant ( JOONIS 1 ) on bensimidasool-pürrolidiin, mis on loodud toimima OX1R ja OX2R kahe antagonistina, pärssides seeläbi oreksiini A ja oreksiin B äratust soodustavat toimet.

Daridoreksandi suukaudne biosaadavus on 62% ja maksimaalne plasmakontsentratsioon saabub 1...2 tunni jooksul (T max ). Daridoreksandi jaotusruumala on 31 liitrit ja see seondub plasmavalkudega 99,7%. Daridoreksant metaboliseerub ulatuslikult, peamiselt CYP3A4 vahendusel (89%). Peamine eritumistee on väljaheitega (57%); väiksem osa (28%) eritub uriiniga, peamiselt metaboliitidena. Daridoreksandi lõplik poolväärtusaeg on ligikaudu 8 tundi. Daridoreksandi kiire imendumine uinumise korral, samuti selle kliirensi profiil, mille tulemuseks on 80% eliminatsioon pärast öist und, võivad jääkmõjusid minimeerida.
Kõrvaltoimed ja ravimite koostoimed 5.6 : Kliinilistes uuringutes (>5% ja rohkem kui platseeboga) olid kõige sagedasemad daridoreksandiga seotud kõrvaltoimed peavalu ja unisus või väsimus. Toote etikett sisaldab ka hoiatust süveneva depressiooni ja enesetapumõtete ning keeruliste unekäitumiste, sealhulgas unes kõndimise, unes autojuhtimise ja muude tegevustega, kui inimene ei ole täielikult ärkvel (nt toidu valmistamine/söömine, helistamine, seks) eest. . Patsiendid peavad nende kõrvaltoimete ilmnemisel viivitamatult ühendust võtma oma tervishoiuteenuse osutajaga. Kui patsient uinub või ärkab, võivad tekkida hallutsinatsioonid ja unehalvatus (ajutine võimetus liikuda või rääkida), mis kestavad kuni mitu minutit. Daridoreksant on narkolepsiaga patsientidele vastunäidustatud.
Puuduvad andmed daridoreksandi kasutamise kohta rasedatel naistel, et teha kindlaks ravimiga seotud suurte sünnidefektide, raseduse katkemise või muude ema või loote ebasoodsate tagajärgede riski. Loomade reproduktiivsusuuringutes ei põhjustanud daridoreksant lootetoksilisust annustes, mis olid kuni 8-10 korda suuremad kui maksimaalne soovitatav annus inimesele. Puuduvad andmed daridoreksandi esinemise kohta inimese rinnapiima, toime kohta rinnaga toidetavale imikule või piimatoodangule, kuid ravimit ja selle metaboliite tuvastati lakteerivate näriliste piimas. Seetõttu tuleb imikuid, kes võivad rinnapiima kaudu daridoreksandiga kokku puutuda, jälgida liigse sedatsiooni suhtes. Daridoreksant on föderaalselt kontrollitav aine, kuna seda võib kuritarvitada ja see võib põhjustada sõltuvust.
Annustamine ja manustamine 5.6 : Daridoreksanti turustatakse suukaudseks manustamiseks mõeldud 25 mg ja 50 mg õhukese polümeerikattega tablettidena. Soovitatav annus on 25 mg kuni 50 mg üks kord öösel, võttes 30 minuti jooksul enne magamaminekut ja vähemalt 7 tundi enne planeeritud ärkamist. Uneaeg võib viibida, kui daridoreksanti võetakse söögi ajal või vahetult pärast seda. Daridoreksanti ei tohi võtta koos teiste kesknärvisüsteemi depressantide ega alkoholiga. Patsiente tuleb ka soovitada vältida autojuhtimist, raskete masinate käsitsemist või mis tahes tegevust, mis nõuab selget mõtlemist, samal ajal kui ravimi mõju kogeb. Mõõduka maksakahjustusega patsientidel on maksimaalne soovitatav annus 25 mg mitte rohkem kui üks kord ööpäevas. Daridoreksanti ei soovitata kasutada raske maksakahjustusega patsientidel. Daridoreksandi samaaegset kasutamist tugevate CYP3A4 inhibiitoritega tuleb vältida ning annust tuleb piirata 25 mg-ni patsientidel, kes saavad samaaegset ravi mõõduka CYP3A4 inhibiitoritega. Samuti tuleb vältida daridoreksandi samaaegset kasutamist mõõduka kuni tugeva toimega CYP3A4 indutseerijatega.
Aducanumab-avwa (Aduhelm, Biogen)
Näidustused ja kliiniline profiil 7.8 : Aducanumab-avwa on amüloid-beeta (AB) suunatud antikeha, mis on näidustatud Alzheimeri tõve raviks ja see on esimene uus selle haiguse ravimeetod alates 2003. aastast. Heakskiit anti kiirendatud heakskiitmisprotsessi alusel, mida kasutati ravimite jaoks, mis annavad tähendusliku ravi. terapeutiline eelis võrreldes olemasolevate ravimeetoditega raske või eluohtliku haiguse korral. Selle näidustuse jätkuv heakskiitmine sõltub kliinilise kasu kontrollimisest kinnitavates uuringutes. Alzheimeri tõbi, pöördumatu, kurnav ja progresseeruv ajuhäire, mis mõjutab 6,2 miljonit ameeriklast, õõnestab aeglaselt mälu, mõtlemist ja lõpuks ka võimet täita isegi lihtsaid ülesandeid. Kuigi Alzheimeri tõve spetsiifiline põhjus pole täielikult teada, iseloomustavad Alzheimeri tõbe aju muutused, sealhulgas amüloidnaastud ja neurofibrillaarsed (tau) puntrad, mille tulemuseks on mälu ja mõtlemist reguleerivate kriitiliste ajupiirkondade neuronite kadu. Praegu saadaval olevad ravimeetodid ravivad ainult haiguse sümptomeid, kuid aducanumab-avwa on esimene aine, mis on suunatud Alzheimeri tõve oletatavale protsessile ja muudab seda.
Aducanumab-avwa kiirendatud heakskiitmine põhines kolme topeltpimeda, platseebokontrolliga, annusevahemikuga uuringu tulemustel, mis hõlmasid kokku 3482 patsienti, kellel oli Alzheimeri tõve kerge kognitiivne häire või kerge dementsuse staadium. Uuringutes, mida tuntakse kui EMERGE (uuring 1), ENGAGE (uuring 2) ja PRIME (uuring 3), kasutati positronemissioontomograafiat Alzheimeri tõve patoloogiaga seotud ajupiirkondade AB naastude kvantifitseerimiseks võrreldes haigusega mitteseotud ajupiirkondadega. Patsientidel, kes said aducanumab-avwat, vähenes AB-naastu märkimisväärne annusest ja ajast sõltuv (59%-71% uuringutes), samas kui kontrollidel AB-naastu vähenemist ei täheldatud. Seni puuduvad andmed ohutuse või efektiivsuse kohta aducanumab-avwa-ravi alustamise kohta Alzheimeri tõve varasemates või hilisemates staadiumides.
Farmakoloogia ja farmakokineetika 7.8 : Aducanumab-avwa on inimese immunoglobuliin-gamma (IgG) 1 monoklonaalne antikeha, mis on võimeline ületama hematoentsefaalbarjääri, et selektiivselt sihtida ja siduda agregeerunud lahustuvaid oligomeere ja AB naastude lahustumatuid fibrillide konformatsioone ajus. Biokeemilised uuringud on näidanud, et aducanumab-avwa seondub selektiivselt AB aminohapetest 3 kuni 7 moodustatud lineaarse epitoobiga, minimeerides seeläbi koostoimeid teiste valkudega. Tuginedes nõrgale monovalentsele afiinsusele, kiirele sidumiskineetikale ja tugevale aviidsusele epitoobirikaste agregaatide suhtes, on näidatud, et aducanumab-avwa eristab AB monomeere ja oligomeerseid või fibrillaarseid agregaate.
Aducanumab-avwa püsikontsentratsioon saavutatakse 16-nädalase korduva annustamise järel iga 4-nädalase režiimiga. Maksimaalne kontsentratsioon (C max ), minimaalne kontsentratsioon (C min ) ja plasmakontsentratsiooni ja aja kõvera alune pindala püsikontsentratsiooni faasis suureneb annusega proportsionaalselt standardse annustamisvahemiku (1–10 mg/kg iga 4 nädala järel). Hinnanguline jaotusruumala tasakaaluseisundis on 9,6 l. Sarnaselt endogeense IgG-ga hüdrolüüsitakse aducanumab-avwa valgu kataboolsete radade kaudu väikesteks peptiidideks ja lõpuks aminohapeteks. Aducanumab-avwa terminaalne poolväärtusaeg on umbes 25 päeva. Neeru- või maksakahjustusega patsientide farmakokineetikat hindavatest uuringutest ei ole teatatud, kuid eeldatavasti ei eritu ravim neerude kaudu ega metabolismi maksas. Kehakaalu, vanuse, soo ja rassi erinevused võivad põhjustada ravimi ekspositsiooni muutumist, kuid ei leitud, et see mõjutaks oluliselt kliinilist vastust.
Kõrvaltoimed ja ravimite koostoimed 7.8 : Kõige sagedamini teatatud aducanumab-avwa kõrvaltoimed on amüloidiga seotud kujutise kõrvalekalded (ARIA); peavalu; langeb; kõhulahtisus; ja segasus, desorientatsioon ja muutunud vaimne seisund. Toote etiketil on hoiatus ARIA eest, mis avaldub enamasti ajuosade ajutise tursena; see toime taandub tavaliselt aja jooksul ega põhjusta teadaolevalt sümptomeid. Mõned ARIA-ga uuringus osalenud patsiendid teatasid siiski peavalust, segasusest, pearinglusest, nägemise muutustest või iiveldusest, mis on tingitud aducanumab-avwast. Harva on aducanumab-avwa kasutamist seostatud ülitundlikkusreaktsioonidega, sealhulgas angioödeemi ja urtikaariaga.
Annustamine ja manustamine 7.8 : Aducanumab-avwa tarnitakse säilitusainetevaba, steriilse IV süstimise lahusena üheannuselistes viaalides 170 mg/1,7 ml (100 mg/ml) ja 300 mg/3 ml (100 mg/ml). Pärast esialgset tiitrimist (üle 6 nädala) on soovitatav annus 10 mg/kg IV infusioonina ligikaudu 1 tunni jooksul iga 4 nädala järel, vähemalt 21-päevase vahega. Esialgne tiitrimine hõlmab 1. ja 2. infusiooni puhul 1 mg/kg annuste manustamist; 3. ja 4. infusiooni puhul annused 3 mg/kg; ja 6 mg/kg annused 5. ja 6. infusioonide puhul. Annuse kohandamine neeru- või maksakahjustuse korral ei ole ette nähtud.
Otesekonasool (Vivjoa, Mycovia)
Näidustused ja kliiniline profiil 9.10 : Otesekonasool on esimene ja ainus ravim, mis on spetsiaalselt näidustatud korduva vulvovaginaalse kandidoosi (RVVC) esinemissageduse vähendamiseks naistel, kellel on anamneesis RVVC ja kellel ei ole paljunemisvõimet. Selle kasutamine on vastunäidustatud rasedatele ja imetavatele naistele. RVVC, üldtuntud kui krooniline pärmseene infektsioon, on defineeritud kui kolm või enam sümptomaatilist ägedat pärmseene infektsiooni episoodi 12-kuulise perioodi jooksul. Sümptomiteks on tupe sügelus, põletustunne, ärritus ja põletik ning mõned patsiendid kogevad ebanormaalset tupevoolust ja valulikku urineerimist või seksuaalvahekorda. Arvatakse, et 75% täiskasvanud naistest põeb elu jooksul vähemalt ühe pärmseene infektsiooni ja ligikaudu 50% kogeb kordumist; Nendest naistest areneb RVVC välja kuni 90%-l. FDA andis oteseconazole Fast-Track ja Qualified Infectious Disease Product nimetused.
Otesekonasool kiideti heaks kolme III faasi kliinilise uuringu efektiivsusandmete põhjal, milles osales 875 patsienti 232 kohast 11 riigis. Kahes ülemaailmses VIOLET-uuringus ei esinenud 93–96% RVVC-ga naistest, kes said otesekonasooli, 48-nädalase säilitusperioodi jooksul haiguse kordumist; seevastu 57–61% platseebo saajatest ei esinenud kordumist. USA-s läbiviidud uuringus UltraVIOLET paranes 89,7% RVVC-ga naistest, kes said otesekonasooli, oma esialgne pärmseene infektsioon ja neil ei olnud 50-nädalase säilitusperioodi jooksul kordumist, samas kui 57,1% flukonasooli ja platseebot saanud naistest ei kordunud.
Farmakoloogia ja farmakokineetika 9.10 : Otesekonasool ( JOONIS 2 ) on uudne tetrasooli seenevastane derivaat, mis inhibeerib 14-alfa-demetülaasi (CYP51), seeneensüümi, mis katalüüsib olulise seente steroolergosterooli biosünteesi varajast etappi. Lisaks põhjustab 14-alfa-demetülaasi inhibeerimine 14-metüülitud steroolide akumuleerumist, mis on seentele toksilised. Tetrasooli metalli siduva rühma kaasamise tõttu selle struktuuri on otesekonasoolil vähenenud afiinsus inimese CYP ensüümide suhtes ning eeldatavasti paremad kõrvaltoimete ja ravimite koostoimete profiilid.

Otesekonasooli maksimaalse plasmakontsentratsiooni saavutamise aeg on vahemikus 5...10 tundi. Manustamine koos kõrge rasvasisaldusega ja kõrge kalorsusega toiduga suurendas C max ja AUC 0-72h vastavalt 45% ja 36%, kuid madala rasvasisaldusega ja madala kalorsusega eine puhul olulisi erinevusi ei täheldatud. Otesekonasooli jaotusruumala on ligikaudu 423 l ja ravim seondub >99% plasmavalkudega. Loomkatsed näitavad, et otesekonasooli ekspositsioon tupe kudedes on võrreldav plasma ekspositsiooniga. Otesekonasool ei metaboliseeru olulisel määral. Ligikaudu 56% suukaudsest annusest eritub väljaheitega sapiga ja 26% eritub uriiniga. Otesekonasooli keskmine poolväärtusaeg on ligikaudu 138 päeva.
Kõrvaltoimed ja ravimite koostoimed 9.10 : Kõige sagedamini teatatud kõrvaltoimed olid peavalu ja iiveldus, mida esines vastavalt 7,4% ja 3,6% uuringus osalejatest. Kliinilistes uuringutes <2% patsientidest teatatud kõrvaltoimete hulka kuulusid düspepsia, kuumahood, düsuuria, reproduktiivsüsteemi häired (menorraagia, metrorraagia, vulvovaginaalne ärritus) ja vere kreatiinfosfokinaasi taseme tõus. Loomkatsetes leiti, et otesekonasool kahjustab looteid; seetõttu on see vastunäidustatud reproduktiivse potentsiaaliga naistele, samuti rasedatele ja imetavatele naistele, kuna see võib ohustada lootele või rinnaga toidetavale imikule. Otesekonasool on vastunäidustatud ka patsientidele, kellel on teadaolev ülitundlikkus ravimi suhtes.
Otesekonasool on rinnavähi resistentsuse valgu (BCRP) inhibiitor ja samaaegne kasutamine BCRP substraatidega, nagu rosuvastatiin, võib suurendada substraadi kokkupuudet, suurendades nende ainetega seotud kõrvaltoimete riski. Seetõttu on soovitatav kasutada BCRP substraadi väikseimaid efektiivseid alg- ja säilitusannuseid, kui seda manustatakse samaaegselt otesekonasooliga, ning patsienti tuleb jälgida kõrvaltoimete suhtes.
Annustamine ja manustamine 9.10 : Otesekonasool on saadaval suukaudseks manustamiseks mõeldud 150 mg kõvade želatiinkapslitena. Soovitatavaid annustamisskeemi on kaks: 1) ainult otesekonasool ja 2) otesekonasool pluss flukonasool. Toote etiketil on nende režiimide jaoks üksikasjalikud annustamisskeemid. Kerge kuni mõõduka neerukahjustusega või kerge maksakahjustusega patsientidel ei soovitata annust kohandada; Otesekonasooli ei soovitata siiski kasutada raske neerukahjustusega või lõppstaadiumis neeruhaigusega (dialüüsiga või ilma) või mõõduka või raske maksakahjustusega patsientidel.
VIITED
1. Adbry (tralokinumab-ldrm) pakendi infoleht. Madison, NJ: LEO Pharma Inc; juuli 2022.
2. Wollenberg A, Howell MD, Guttman-Yassky E, et al. Atoopilise dermatiidi ravi tralokinumabiga, anti-IL-13 mAb. J Allergy Clin Immunol . 2019; 143 (1): 135–141.
3. Saphnelo (anifrolumab-fnia) pakendi infoleht. Wilmington, DE: AstraZeneca Pharmaceuticals LP; juuli 2022.
4. Anifrolumab: teave ravimi kohta. Ajakohane. Waltham, MA: UpToDate; 2022. www.uptodate.com. Accessed June 28, 2022.
5. Quviviq (daridoreksant) pakendi infoleht. Radnor, PA: Idorsia Pharmaceuticals US Inc; aprill 2022.
6. Markham A. Daridorexant: esimene heakskiit. Narkootikumid . 2022;82(5):601-607.
7. Aduhelm (aducanumab-avwa) pakendi infoleht. Cambridge, MA: Biogen Inc; juuni 2021.
8. Sevigny J, Chiao P, Williams L jt. Aducanumab (BIIB037), amüloid-beeta monoklonaalne antikeha, prodromaalse või kerge Alzheimeri tõvega patsientidel: randomiseeritud topeltpime platseebokontrollitud faasi 1b uuringu vahetulemused. Alz dementsus . 2015;11 (7 lisa 6): P277.
9. Vivjoa (oteseconazole) pakendi infoleht. Durham, NC: Mycovia Pharmaceuticals, Inc; aprill 2022.
10. Sobel JD, Nyirjesy P. Oteseconazole: edusammud korduva vulvovaginaalse kandidoosi ravis. Tuleviku mikrobiol . 2021;16(18):1453-1461.
Selles artiklis sisalduv sisu on mõeldud ainult informatiivsel eesmärgil. Sisu ei ole mõeldud professionaalsete nõuannete asendamiseks. Selles artiklis esitatud teabele tuginemine on ainult teie enda riisikol.











